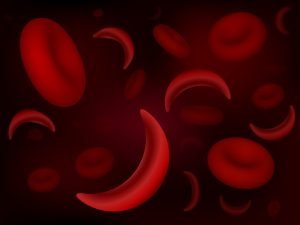
مرض وراثي يصيب خلايا الدم الحمراء، حيث يفتقر الدم لوجود خلايا دم حمراء صحّية كافية لحَمل الأكسجين في الجسم كلّه.
تتحرّك خلايا الدم الحمراء المرِنة والمستديرة بسهولة في الأوعية الدمويّة، لكن عند الإصابة بفقر الدم المِنجليّ؛ تتشكّل خلايا الدم الحمراء على شكل مِنجل أو هلال.
قد تلتصق هذه الخلايا اللزجة والقاسية في الأوعية الدمويّة الصغيرة؛ ممّا قد يبطّئ أو يمنع تدفّق الدم والأكسجين إلى أجزاء الجسم.
تظهر عادة أعراض المرض عند 5 أشهر من العمر، ولكن قد تتغيّر مع مرور الوقت وتختلف من شخص لآخر، وقد تتضمّن:
1. فقر الدم:
تموت الخلايا المِنجليّة وتتفتّت بسهولة، ممّا يقلّل عدد خلايا الدم الحمراء.
2. نوبات الألم:
من الأعراض الرئيسة لفقر الدم المنجليّ، ويحدث عندما تمنع خلايا الدم الحمراء المنجليّة تدفّق الدم عبر الأوعية الدمويّة إلى أعضاء الجسم.
قد يحدث الألم أيضًا في العظام.
3. تورّم الكفّين والقدمَين:
يحدث التورّم بسبب خلايا الدم الحمراء المنجليّة التي تمنع تدفّق الدم إلى اليدَين والقدَمين.
4. حالات عدوى متكرّرة:
هناك احتمال أن تُتلف الخلايا المنجليّة الطحال، ممّا يجعل المصاب أكثر عُرضة للإصابة بالعدوى.
5. تأخُّر النموّ أو تأخّر البلوغ:
تزوّد خلايا الدم الحمراء الجسم بالأكسجين والعناصر المغذّية اللازمة للنموّ، ويمكن لنقص عدد خلايا الدم الحمراء السليمة أن يبطّئ نموّ الرضع والأطفال ويؤخّر سنّ البلوغ لدى المراهقين.
6. مشاكل الإبصار والرؤية:
يمكن أن تسدّ الخلايا المنجليّة الأوعية الدمويّة الصغيرة التي تغذّي العينَين، ممّا يؤدّي إلى إتلاف الشبكيّة ومشاكل في الرؤية.
يحدث المرض عند عمليّة تشكيل الهيموغلوبين أو خضاب الدم، والذي يحصل على حمض نوويّ ريبوزيّ رسول حيث يحل GUG مكان GAG بالتالي يحلّ الفالين محلّ حمض الغلوتاميكفي.
ممّا يجعل الروابط الهيدروجينية التي تشكّل البُنى الأعلى من الهيموغلوبين تأخذ شكلاً غير طبيعيّ للبروتين المتشكّل.
ويعدّ جهاز VARIANT المقدّم من شركة بيوراد من أهمّ الأجهزة في العالم لفَحص هذا المرض.
يمكن تشخيص المرض بواسطة فحص مِخبريّ يحدّد خضاب الدم المنجليّ.
يعدّ تشخيص حاملِي المرض مهمّاً جداً، خاصة في العائلات التي يمكن أن تكون حاملة لمورثة Beta- thalassemia.
حيث إنّ اتّحاد المورّثتَين المعطوبتَين مع سلاسل بيتا جلوبين لدى الأحفاد قد يؤدّي إلى مرض خطير، يشمل أعراض المرضَين معا.
وتُتيح فحوصات الحمض النوويّ الريبيّ المنزوع الأكسجين وفحص تشخيص الجنين في مراحل مبكّرة من الحمل، تِبيان ما إذا كان الجنين معرّضاً للإصابة بهذا المرض الخطير.
1. التسريب:
إعطاء سائل أو محلول علاجيّ في الوريد أو المضادّات الحيويّة حسب الحاجة.
2. استبدال الكريات الحمراء:
يتمّ استبدال نِصف كمّية الكريات الحمراء في جسم المريض، بشكل تدريجيّ، بكُريات حمراء سليمة.
3. علاج وقائيّ ممكن بواسطة دواء سامّ للخلايا:
يساهم في تحسين مُجمل الأعراض السريريّة، ولكن هذا العلاج ليس ملائماً لجميع المرضى، بل لعدد محدود فقط.
يمكن الوقاية من هذا المرض مبكّراً بإجراء فحوصات ما قبل الزواج، لمعرفة إذا ما كان أحد الطرفين حاملاً للمرض، فهذه الطريقة سهلة ويسيرة ومتوفّرة في معظم المختبرات الطبّية.
هناك العديد من المسمّيات لهذا المرض ومنها:
1. مرض الهيموغلوبين SS:
تحدث عند وراثة الشخص نسخاً من جين الهيموغلوبينS من كلا والديه، ويمثّل أكثر أنواع فقر الدم المنجليّ انتشاراً وشدّة، إذ يعاني الأفراد المصابون بهذا المرض أسوأ الأعراض وبنِسَب أعلى مقارنةً بالأنواع الأخرى.
2. مرض الهيموغلوبين SC:
تحدث هذه الحالة عند وراثة الشخص جين الهيموغلوبين C من أحد الوالدين، والهيموغلوبينS من الآخر، وتُمثّل هذه الحالة ثاني أكثر أنواع فقر الدم المنجليّ شيوعاً.
يعاني الأفراد المصابون بهذا النوع من أعراض مشابِهة لتلك التي يعاني منها المرضى المصابين بمرض الهيموغلوبين SS، إلّا أنّ فقر الدم يكون أقلّ شدّة.
3. مرض الهيموجلوبين SB+ -بيتا- الثلاسيمي:
يؤثّر الطفرة في هذه الحالة على إنتاج جين بيتاغلوبين، ويعاني المريض في هذه الحالة من انخفاض حجم خلايا الدم الحمراء نظراً لانخفاض كمّية بروتين البيتا الذي يتمّ إنتاجه.
في حال وراثة الشخص جين الهيموغلوبينS فسيعاني الشخص من ثلاسيميا الهيموجلوبين S بيتا، وفي هذه الحالة تكون الأعراض غير شديدة.
4. مرض الهيموجلوبين SB 0 بيتا- صفر الثلاسيمي:
يؤثّر المرض في هذه الحالة في جين البيتاغلوبين، حيث تكون الأعراض مشابهة لأعراض مرض الهيموغلوبينSS ، ويُشار إلى أنّ أعراض مرض الهيموجلوبين SB 0 -بيتا-صفر- الثلاسيمي تكون أكثر شدّة في بعض الأحيان، وقد تتطوّر هذه الحالة بشكل سيء.
أنواع أخرى: مرض الهيموغلوبين SD، ومرض الهيموغلوبين SE، مرض الهيموغلوبين SO.
تمثّل هذه أنواع فقر الدم المنجليّ الأكثر نُدرة وعادةً لا تظهر أيّ أعراض شديدة على المريض.
في عام 1846 يعتقد أنّه تمّ اكتشاف أوّل حالة من مرض فقر الدم المنجليّ حيث كانت النتائج التشريحيّة الرئيسيّة تشير إلى غياب الطحال، وكانت هناك أيضاً تقارير بين العبيد الأفارقة في الولايات المتّحدة واظهار مقاومة للملاريا.
الطفرات الغير طبيعية في خلايا الدم الحمراء وُصفت لأول مرّة من قِبل إرنست إدوارد الحديد 1877-1959، المتدرّب وأستاذ الطبّ جيمس ب. هيريك 1861-1954، في عام 1910.
لاحظ الحديد أنّ الخلايا الدمويّة في دم رجلٍ يُدعى والتر كليمنت نويل قد أخذت شكل مِنجل، في السنة الأولى وهو طالب يبلغ من العمر 20 عاماً.
بعد فترة وجيزة من تقرير هيريك، ظهرت قضية أخرى في ولاية فرجينيا، تتحدّث عن مريض أُدخل إلى مستشفى جامعة فرجينيا في 15 نوفمبر، 1910 مصاب بنفس الطفرة السابقة.
لاحقاً، استُخدم اسم “فقر الدم المنجلي” لأوّل مرّة عام 1922 عن طريق فيرن ميسون.
تمّ إدخال دراسات التاريخ الطبيعيّ على نطاق واسع وإجراء المزيد من الدراسات في 1970 و1980، ممّا أدى إلى زيادة انتشار استخدام العلاج الوقائي ضدّ الالتهابات.
في عام 1972، تمّ إصدار فيلم تلفزيونيّ يُدعى “أصدقاء على الشاطىء” وقد حصل على جائزة الإيمي. يصوّر الفيلم قصّة والدَي طفل يعاني من مرض “هيدروكسي كاربامايد” أو الخلية المنجلية.
تتعدّد الأبحاث و الاكتشافات المرتبطة بمرض فقر الدم المنجليّ، حيث توصّل مجموعة من الباحثين إلى اكتشاف علاج جيني رائد لمرض الخلية المنجليّة وقد حقق نتائج جيّدة ومبشّرة.
حين استُخدم مع صبيّ فرنسيّ في الثالثة عشرة من عمره يعاني من مرض وراثيّ في الدم.
وتعدّ هذه الحالة الأولى لمرض فقر الدم المنجليّ التي يتمّ علاجها بالتكنولوجيا التي تنتجها شركة “بلوبيرد بيو”، التي تعرف باسم “لنتيجلوبن بي بي.305”.
حيث تعتقد الشركة الأمريكية المعنيّة بالتكنولوجيا الحيويّة أنّ هذه الطريقة يمكن أن تمثّل علاجاً للمرض.
يعتمد العلاج الجيني الذي تقدّمه “بلوبيرد” على علاج الحالات من خلال استخراج الخلايا الجذعيّة للدم ثمّ إضافة نسخة سليمة للجين المَعيب.
وأظهرت النتائج أنّ المريض الفرنسيّ ظلّ دون حاجة إلى عمليّة نقل دم حفاظاً لأكثر من ثلاثة أشهر وأنّ جسمه أنتج 45% ممّا يعرف بالهيموجلوبين المضادّ للخلية المنجليّة خلال ستّة أشهر.
ويشهد العلاج الجينيّ حالياً طفرة جديدةً بعد سلسلة من الانتكاسات أواخر التسعينات من القرن الماضي وأوائل القرن الحادي والعشرين وتستثمر شركات دواء كبرى الكثير في هذا المجال.